Protein structure and dynamics
Map the conformational landscape of protein folding and complex formation
Bridge the gap between static structures and dynamic function with single-molecule clarity
Proteins are highly dynamic – undergoing conformational changes during folding, ligand binding, and higher-order complex formation. Identifying conformational states and tracking interconversions between them is crucial to understanding how protein structure and dynamics relate to their function. Mapping protein misfolding and aggregation is also vitally important for understanding their role in disease.
However, catching these transient or early events is often challenging with static or ensemble techniques alone. By tracking individual molecules in real time, researchers can measure complex kinetics, map dynamic conformational shifts with high temporal resolution and understand the stoichiometries of complex formation.
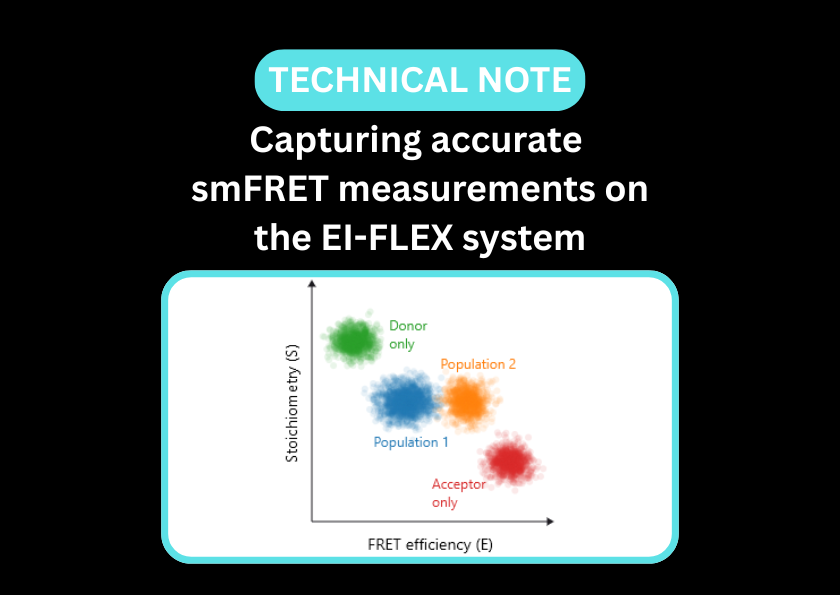
Single-molecule Förster resonance energy transfer (smFRET) and Fluorescence correlation spectroscopy (FCS) bring static snapshots to life. By measuring distances between specific residues in real time, smFRET maps the conformational landscape of a protein, revealing how it shifts between active, inactive, and intermediate states. This is particularly transformative for intrinsically disordered proteins (IDPs), offering complementary insights to traditional structural biology techniques. Additionally, FCS monitors the protein’s hydrodynamic size and assembly state, allowing for the precise tracking of folding pathways, allosteric transitions, and the earliest stages of aggregation.
Transform static snapshots into dynamic structures in their native environments
- Capture large-scale domain movements of proteins, such as to identify transient allosteric pockets for novel small-molecule binders
- Map how conformational states shift in response to ligand binding, facilitating the distinction between induced-fit and conformational selection mechanisms
- Define the 'structural cloud' of IDPs to identify specific motifs that can be stabilised or inhibited by drug candidates
- Quantify distinct structural sub-populations within a single sample - essential for identifying misfolded or transient species that traditional bulk methods average out
- Monitor how proteins fold or unfold in response to environmental stressors or post-translational modifications
- Utilise F(C)CS to identify soluble, low-order oligomers (the often-toxic precursors to visible aggregates), as well as large-scale fibres
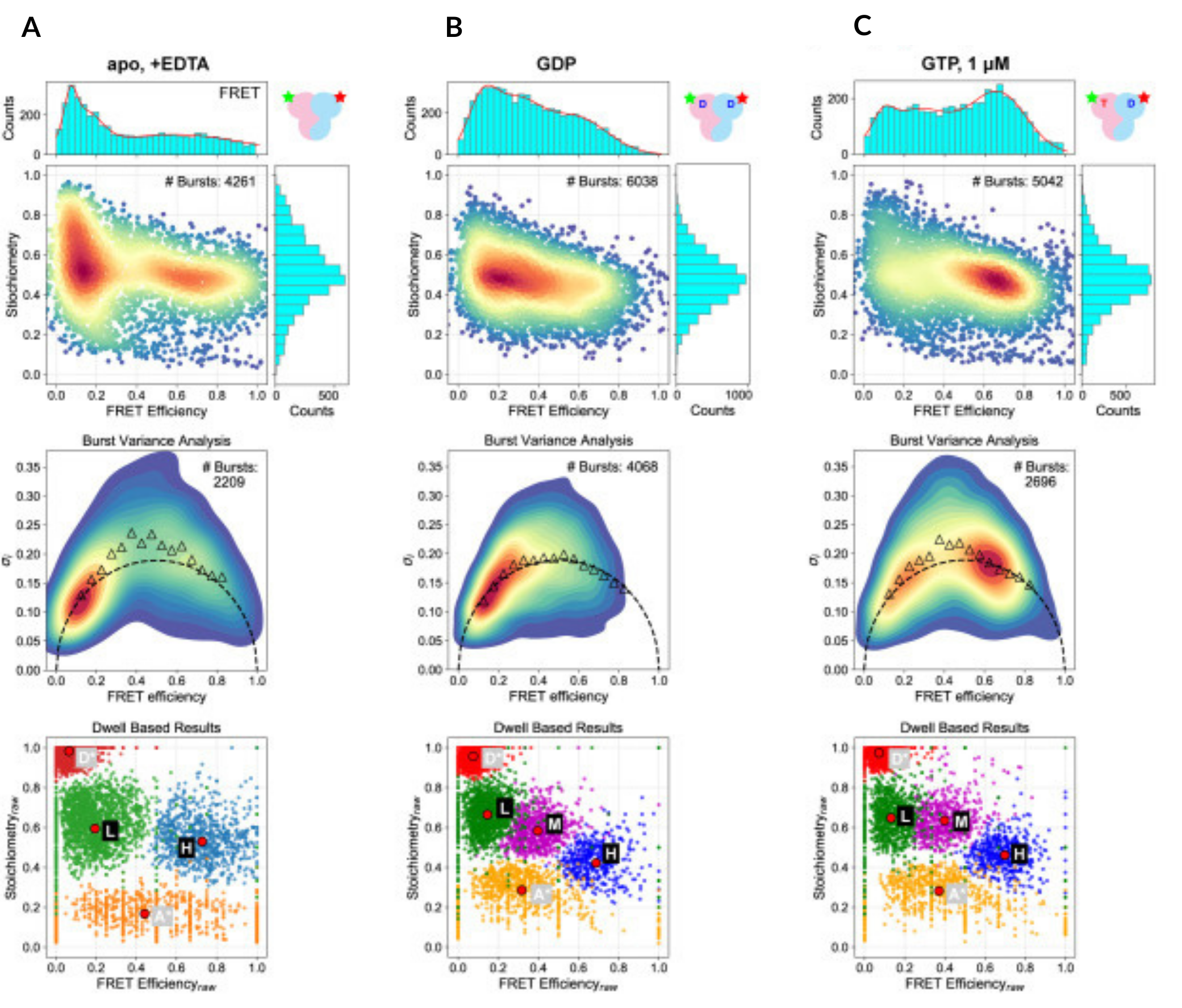
smFRET captures global conformational changes in Rag GTPase upon guanine nucleotide binding
In the absence of guanine nucleotides, Rag GTPase occupies an open, dynamic conformation (low FRET efficiency, left column). This contrasts with dually GDP-bound Rag GTPase, where it remains in an open but static conformation (low FRET efficiency, middle column). When both GDP and GTP are bound, the protein adopts a static closed conformation (high FRET efficiency, right column).
See the paper on this study to explore how smFRET was performed on the EI-FLEX.
Protein structure and dynamics papers and pre-prints featuring EI-FLEX data


Protein structure and dynamics FAQs
How does smFRET complement other structural techniques, such as X-ray crystallography?
While X-ray crystallography provides a high-resolution snapshot of a protein in a crystal lattice, smFRET captures the protein’s behaviour in solution. It can identify multiple interconverting structures and quantify the transition rates between them.
Can smFRET monitor protein folding and unfolding in real-time?
By placing a donor and acceptor at two points on the peptide chain, the distance between them can be measured as the protein folds. This is particularly powerful for identifying short-lived folding intermediates that are the precursors to functional or pathological states.
What can FCS tell us about Intrinsically Disordered Proteins (IDPs)?
IDPs do not have a stable 3D structure, making them challenging to understand with common structural methods. FCS can measure their hydrodynamic radius, which tells us how compact the disordered chain is. By changing salt or temperature, FCS can monitor the collapse of an IDP into a more globular form, a key step in many signalling pathways. FCS also performs well in physiologically relevant media, which is often crucial for working with IDPs.
How do I detect flexibility within a protein?
This is done using Burst Variance Analysis (BVA) in smFRET. If a protein is rigid, the FRET signal variance is purely due to light physics (shot noise). If the protein is flexible or ‘breathing’, the variance will be significantly higher.
Can smFRET study protein misfolding and Prion-like behaviour?
smFRET is an ideal tool for studying neurodegenerative diseases. You can watch a single protein molecule transition from its healthy fold into a misfolded state that is prone to aggregation. Because it’s a single-molecule technique, you can catch the first molecule that misfolds before it triggers a chain reaction.
Can FCS track the early stages of protein oligomerisation?
As proteins begin to clump together (forming dimers, trimers, etc.), two things happen in FCS: the diffusion slows down, and the molecular brightness increases. If a dimer forms, the particle will be twice as bright as a monomer. This is an ideal technique for studying the early seeds of protein aggregates.
How does protonation or pH affect protein dynamics?
Many proteins act as pH-sensors. By using smFRET, you can monitor how a protein’s structure shifts between different states as you titrate the pH. This is vital for studying proteins that function in acidic environments, like lysosomes or the stomach.
Can I see domain swapping or hinge movements?
Yes. Many proteins consist of multiple domains connected by flexible hinges. By placing the FRET pair across the hinge, you can measure the angular distribution of the domains and how they are influenced by the binding of substrates or cofactors.
Can I investigate how molecular chaperones affect protein dynamics?
smFRET can capture how chaperones induce conformational shifts in target proteins. This is typically observed as a shift from a dynamic, broad FRET distribution of the unfolded protein to a narrow peak as a stable folded structure is induced. FCCS can also directly confirm whether a chaperone has bound a protein.
Can I use smFRET to study allostery?
smFRET can show that binding a ligand at one site changes the structural dynamics at another distant location, such as an active site. This allows you to map the allosteric pathways that proteins use to regulate their own activity or track the allosteric effects of drugs.
“Solution-based measurements, like those taken using the EI-FLEX, avoid surface tethering, which can introduce artefacts, especially for proteins that don’t behave well near hydrophobic surfaces. Keeping proteins in solution lets you observe their native conformation, and it’s much easier experimentally: you label, dilute, and measure, often getting results in minutes rather than days or weeks with TIRF-based systems."
Associate Professor Kuang Shen, UMass Chan

Other resources you might be interested in