The success of structural biology
For decades, structural biology has been defined by the quest for the ‘perfect snapshot’. The Protein Data Bank (PDB) stands as a monumental testament to this effort, housing over 250,000 structures that have mapped the blueprints of life with breathtaking precision. From the early days of X-ray crystallography to the resolution revolution of Cryo-EM and the predictive power of AlphaFold, we have become masters at visualising what proteins and other macromolecules look like.
However, for the modern structural biochemist, a high-resolution coordinate set is often just the beginning of the story.
How can we bring these structures to life?
The fundamental paradox of structural biology is that while we solve structures in static states (often trapped in a crystal lattice or frozen in vitreous ice), biology happens in motion. Biomolecules, such as proteins, are dynamic ensembles; they breathe, shift, and undergo transient conformational changes that are often the true drivers of catalysis, signalling, and drug binding.
The challenge we face today isn’t just knowing the ‘A’ and ‘B’ states, but understanding the pathway between them:
- How long does the protein spend in an ‘open’ vs. ‘closed’ state?
- Are there hidden intermediates that only exist for milliseconds?
- How does the local environment (salt concentration, pH, or ligand binding, etc.) shift this equilibrium?
Introducing single-molecule spectroscopy
To bring these snapshots to life, we need tools that can visualise molecules as they move freely in solution. Single-molecule FRET (smFRET) and Fluorescence Correlation Spectroscopy (FCS/FCCS) allow researchers to observe individual molecules as they diffuse through a confocal volume. Unlike ensemble methods that average out the noise, these techniques capture the heterogeneity of biomolecules within a sample; it quantifies the full distribution of conformations, revealing sub-populations and transient states that are invisible to traditional structural techniques.
In this article, we will explore how these techniques can act as the dynamic validator for structures identified by traditional or AI-based techniques, adding a vital complementary technique to current structural toolkits and bringing atomic detail to life by mapping the structural landscapes that govern molecular function.
Defining the vital role of dynamics in structural biology
Understanding how biomolecular structures translate to their function requires dynamics as an additional layer of information. Below, we explore some key biological scenarios where capturing biomolecular dynamics reveals vital mechanistic information that would be missed using static methodologies alone.
- Mapping allosteric networks
Allostery is the phenomenon where a ligand binding at one site triggers a functional change at a distal site. However, in many cases, the crystal structures of the apo (unbound) and holo (bound) states look nearly identical. Often, an allosteric compound doesn’t cause a massive structural overhaul that is easily captured by X-ray crystallography; instead, it subtly shifts the conformational equilibrium.
Allostery is frequently driven by changes in conformational fluctuations rather than large-scale shifts. smFRET can monitor the population shift between an active-like and inactive-like state in real-time, identifying how an allosteric modulator rebalances the energy landscape to favour one functional state over another. In drug discovery, smFRET can be used to quantify exactly how effectively a compound stabilises a non-functional conformation, even if the binding pocket is far from the active site.
- Resolving ‘dark matter’: From flexible loops to intrinsically disordered proteins (IDPs)
Perhaps the most common frustration in structural biology is the missing density— regions of a map where a loop, a C-terminal tail, or an entire domain is too flexible to be resolved. In many cases, they play critical roles in signalling and recognition; traditional structural methods often try to force these regions into a single average coordinate or, more often, leave them out of the model entirely. This leaves a gap in our understanding of how these regions truly behave in solution.
By placing fluorescent dyes around these flexible motifs, we can characterise the many conformational states that they undertake. We can determine if a loop is truly disordered or whether it is switching between discrete, transiently stable states that are too short-lived for other structural techniques to categorise.
- Targeting undruggable proteins
Many high-value targets are deemed undruggable because their static structures lack obvious pockets. However, by monitoring distance distributions across a protein surface with smFRET, we can detect the transient breathing that reveals cryptic pockets and generate a kinetic map. This identifies not just where a pocket might open, but how often and for how long. This data is ideal for determining the feasibility of covalent inhibitors or high-affinity binders against proteins in a specific, fleeting conformation, or for intrinsically disordered proteins.
- Visualising multi-step molecular pathways
Complexes like helicases, polymerases, and transporters operate through a series of coordinated steps. Capturing every possible state using traditional structural techniques is a monumental task, often missing the short-lived intermediates that act as the transition states of the cycle.
Because smFRET observes molecules freely diffusing in solution, it naturally captures these transient intermediates. It allows researchers to build a complete kinetic model of the cycle and map not just what the states look like, but the transition rates between them.
Expanding the structural biology toolkit
X-ray crystallography, Cryo-EM, and NMR are the pillars of the field, providing a wealth of structural information that has driven scientific discovery forward for many years. However, structural biology has always been a game of trade-offs between resolution, size, and environment. smFRET is a highly complementary technique that acts as a powerful bridge, providing the solution-phase context that static or ensemble methods might miss.
Technique | Strengths | smFRET complementarity |
X-Ray Crystallography | Provides unsurpassed atomic precision (<1.5 Å) for well-ordered proteins. | Validates that the crystalline structure matches the conformation found in free solution, removing potential crystal-packing bias. |
Cryo-EM | Captures large, complex assemblies and multiple 3D classes in a near-native state. | Cryo-EM shows the main states, smFRET populates the ‘dark’ intermediates between those classes, revealing the pathways of transition. |
NMR Spectroscopy | Excellent for residue-level dynamics and small-to-midsize proteins in solution. | Offers long-range distance data (up to 10 nm) for large complexes or disordered ensembles that are often too big for NMR or suffer from line-broadening. |
Rather than seeing these techniques as competing, they are most impactful when used as a continuous pipeline:
- Define the Map: Use X-ray Crystallography or Cryo-EM to establish the high-resolution architecture of a protein’s major stable states.
- Sample the Ensemble (Computational Scaffolding): Use AlphaFold to generate high-confidence models of individual domains or complexes. Specialised tools like AlphaFold-Ensemble or Molecular Dynamics (MD) can be used to simulate the potential transition pathways between known structures.
- Validate the Mechanism: This is where smFRET is essential. It provides the experimental temporal resolution to see if the transitions predicted by MD actually occur in solution, at what frequency, and under what physiological conditions (e.g., pH or salt shifts).
By combining these methods, researchers can move from a collection of static structures to a fully realised dynamic pathway, ensuring that structural models are both accurate and biologically relevant.
Beyond the histogram: Adding dynamics to smFRET data
Generating FRET efficiency histograms is a key step during smFRET data analysis. These plots reveal whether samples contain a single population or multiple, heterogeneous species. While this is informative, we need to perform an additional layer of analysis on this data to extract the dynamics.
Burst Variance Analysis (BVA)
In this example, we show two different samples: one contains three DNA oligos with dye pairs placed at different distances apart, while the second sample contains a single DNA hairpin that can open and close. Both of these samples produce FRET efficiency plots with multiple peaks, but the data in this format does not tell us if these are static, heterogeneous populations (DNA oligos) or dynamically interconverting species that are sampling multiple conformations (DNA hairpin).
In this case, burst variance analysis (BVA) is used to quantify changes in the standard deviation of the measured FRET efficiencies beyond what is expected for static molecules, which would suggest the presence of dynamically interconverting species.
By performing BVA analysis on our oligo and hairpin samples, we can see that the FRET efficiencies for the DNA oligo sample do not deviate from the expected standard deviation, indicating that this sample contains three static molecules. Similarly, for the hairpin sample, the constructs with low-FRET and high-FRET values adopt a static conformation. However, constructs that have a mid-FRET efficiency value (indicated by purple triangles) deviate from the expected standard deviation and therefore adopt dynamic conformational changes. This deviation suggests that these molecules undergo transitions between multiple conformational states within the timescale of a single photon burst.
To read more about this application and see how BVA can capture the effect of salt concentration on hairpin conformational dynamics, read our application note here.
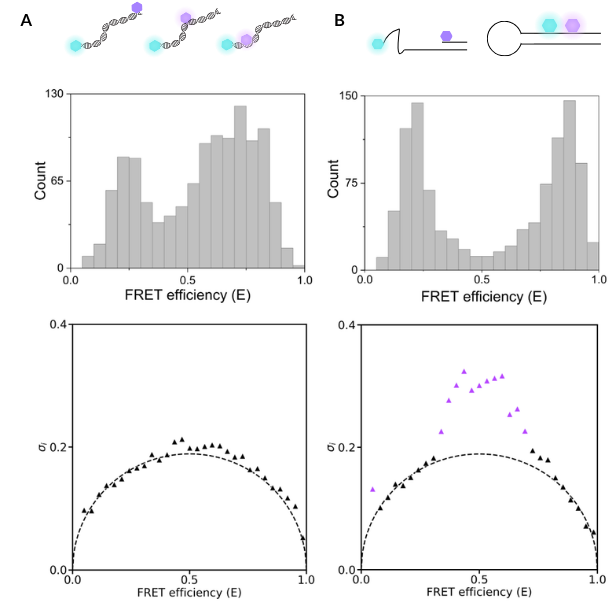
Distinguishing static, heterogeneous populations from dynamically interconverting biomolecules
A static, heterogeneous population (DNA duplexes with different dye pair placements) can produce similar smFRET histograms to a dynamically interconverting population (DNA hairpin), highlighting the importance of additional dynamic analysis of smFRET data.
Hidden Markov Modelling (H2MM)
Another layer of dynamic information comes in the form of Hidden Markov Modelling (H2MM). This process analyses the sequence of individual photon arrivals to infer discrete FRET states and the transition rates between them. In the example below, Howard et al. used smFRET to uncover two intermediate conformations in the bacterial helicase Rep, whereby previous x-ray crystallography data had identified open and closed states1. They used H2MM to investigate the transition rates between these four identified conformations, also exploring the effects of NaCl and DNA on their rates of interconversion.
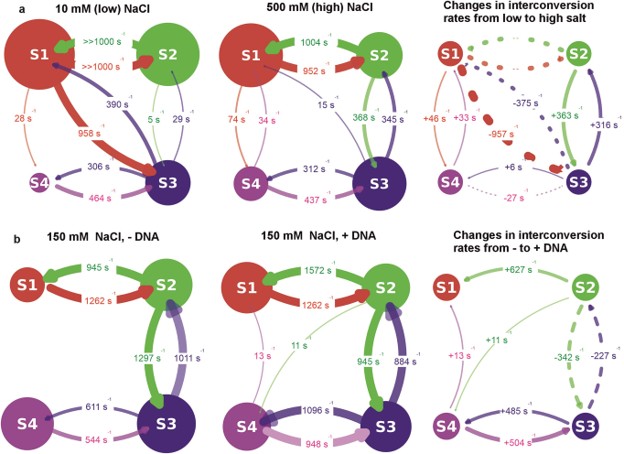
Hidden Markov Modelling can determine interconversion rates of conformations identified by smFRET
H2MM predicts both the sequence that the four distinct conformations of the bacterial helicase Rep undertake, but also the influence of DNA and NaCl on their interconversion rates.
Case Studies
To see the power of smFRET in action, we can look at how it resolves the missing links in both well-folded proteins and disordered ensembles. These examples demonstrate that when structural information is combined with dynamic data, the mechanism becomes clear.
Example 1: Resolving ligand-induced conformational heterogeneity
In this example, Doxsey and Shen used smFRET to resolve the conformational changes of a highly dynamic heterodimeric protein, Rag GTPase, which binds both nucleotides and the serine/threonine protein kinase complex mTORC12.
To explore the conformational changes following guanine nucleotide binding to these subunits, they incubated Rag GTPase with EDTA only (A), GDP only (B), or GTP and GDP (C). They performed smFRET, alongside BVA and H2MM to distinguish dynamic conformational changes from those that are static and to characterise the interconversion between them.
The figure below shows that when guanine nucleotides are absent, Rag GTPase occupies an open conformation (low FRET efficiency), although the protein’s conformation is dynamic due to an increase in the standard deviation of FRET efficiency compared to the theoretically calculated one (as shown by burst variance analysis). This contrasts with dually GDP-bound Rag GTPase, where it remains in an open conformation (low FRET efficiency), but the burst variance analysis indicates that this is a static complex. When both GDP and GTP are bound, the protein adopts a static closed conformation, generating a high FRET state and no deviation from the theoretical standard deviation. Hidden Markov modelling identified a third, mid-FRET state in the nucleotide-bound complexes that was absent in the unbound condition.
The authors calculated the inter-dye distances for the mid- and high-FRET states (61 and 51 Å, respectively), which corroborated with distances determined by crystal structures showing open and closed conformations of the nucleotide-binding domain.
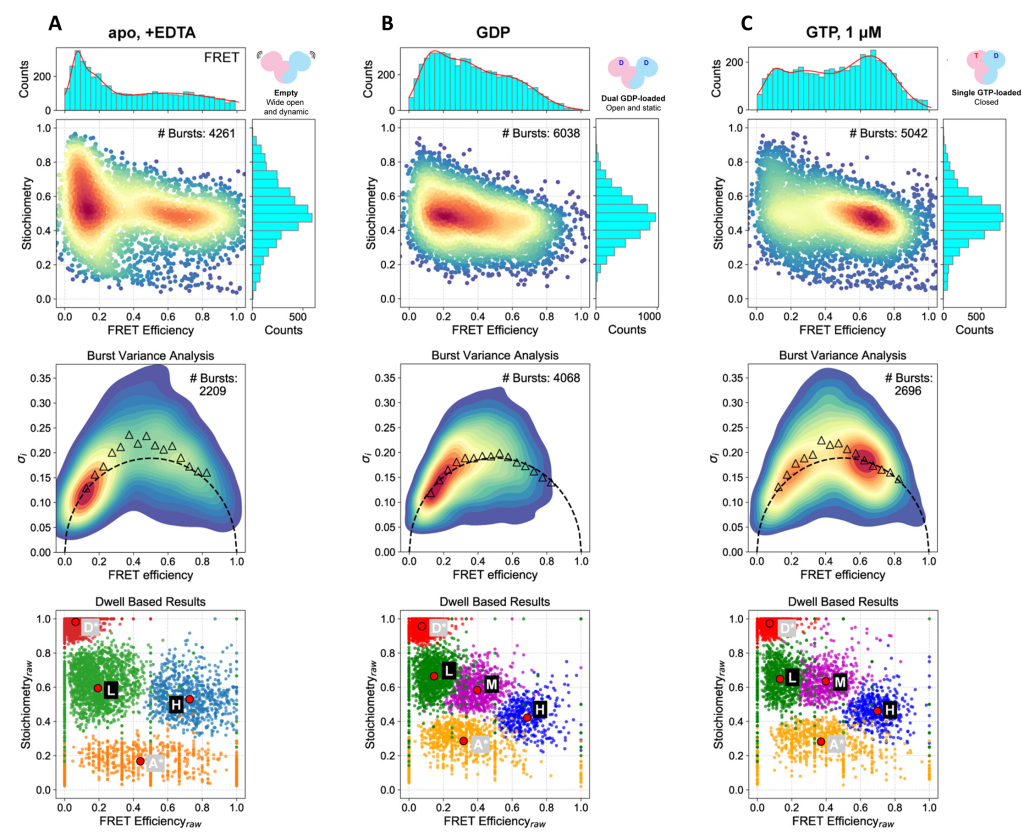
Binding of GDP alone or with GTP induces global conformational changes in Rag GTPase
Top row: smFRET histograms and FRET efficiency vs stoichiometry plots
Middle row: Burst variance analysis, showing theoretical standard deviation of bursts (dotted line) and experimentally observed standard deviation (triangles)
Bottom row: Hidden Markov modelling data indicating donor only (D), acceptor only (A), low-FRET (L), mid-FRET (M) and high-FRET (H) populations
A) Unbound Rag GTPase (EDTA only)
B) Rag GTPase with GDP only
C) Rag GTPase with GDP and GTP
Figure adapted from Doxsey and Shen, 20252
Example 2: Mapping IDP ensemble structures
Naudi-Fabra et al. utilise a powerful integrative approach—combining smFRET, FRET-lifetimes, NMR (chemical shifts and PREs), and SAXS—to quantitatively map the conformational ensemble of the disordered domain of the measles virus phosphoprotein3. When used individually, these techniques typically only capture one aspect of an IDP (like local structure or global size). Therefore, the authors used smFRET to provide critical long-range distance constraints that anchored the entire model.
By incorporating these diverse experimental datasets into the ASTEROIDS algorithm, the team moved beyond simple polymer models, revealing that this viral protein is more compact than a random chain and contains transient, functional structural elements. This study serves as an example of complementarity, demonstrating how smFRET can unify residue-specific NMR data and global SAXS dimensions to produce dynamic conformational ensembles for disordered proteins with high confidence.
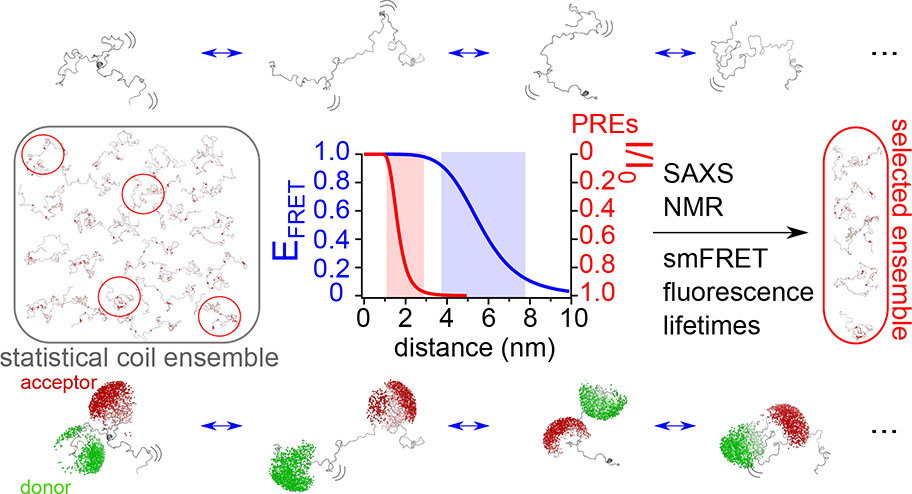
Combining smFRET, SAXS, NMR and lifetime fluorescence to map IDP structures
By combining multiple structural and biophysical techniques with smFRET, a predicted set of conformations undertaken by the IDP measles virus phosphoprotein could be derived.
Conclusion
Structural biology has spent decades perfecting the ‘still life’ of the molecular world. But as our understanding of allostery, intrinsically disordered proteins, and complex catalytic cycles grows, it is becoming clear that these static structures are the beginning of the story, not the end.
The power of smFRET lies in its ability to complement existing biophysical and biochemical protocols. It builds upon the high-resolution detail of X-ray and Cryo-EM, and predictive power of AlphaFold and MD simulations, providing the experimental validation—the temporal resolution—needed to unify these snapshots.
Ultimately, mapping the path from atomic detail to molecular motion provides the necessary context for our structural data, transforming a collection of high-resolution coordinates into a complete mechanical understanding.
References
- Howard, J. A. L. et al. The transitional kinetics between open and closed Rep structures can be tuned by salt via two intermediate states. Nucleic Acids Res. 54, gkaf1483 (2026).
- Doxsey, D. D. & Shen, K. Global conformation of the Rag GTPase heterodimer governs eukaryotic amino acid sensing. Proceedings of the National Academy of Sciences 122, e2517050122 (2025).
- Naudi-Fabra, S., Tengo, M., Jensen, M. R., Blackledge, M. & Milles, S. Quantitative Description of Intrinsically Disordered Proteins Using Single-Molecule FRET, NMR, and SAXS. J. Am. Chem. Soc. 143, 20109–20121 (2021).