The messy middle of biophysics
For decades, the gold standard of biophysics has lived at two extremes. On one side, we have the ‘spherical cow’ approach using purified proteins in optimised, crystal-clear buffers. It’s elegant and precise, but it lacks the soul of biology; the crowding, the competitive binding, and the sheer chaos of a living system. On the other side, we have live-cell imaging: the ultimate truth, but an optical and technical nightmare where phototoxicity, cell movement, and background noise often obscure the very molecules we want to see.
The compromise between the sterile test tube and the living cell offers a messy middle ground, whereby single-molecule biophysics is performed directly in cell lysates and serum.
By analysing biomolecules in these matrices, we preserve their native environment, protecting biologically-relevant factors such as post-translational modifications (PTMs), natural regulators, and macromolecular crowders, while maintaining the optical clarity and experimental control of an in vitro sample. In this environment, single-molecule techniques like smFRET, FCS, and FCCS perform just as well as if these molecules were purified or still within intact cells.
In this article, we’ll take you through how to get the most out of your single-molecule spectroscopy studies, backed up by case studies showing these techniques in action and proving that modern biophysics can thrive with ‘messy’, physiologically relevant samples.
Preserving biomolecular authenticity with in-cell labelling
The primary challenge of biophysics in crude media is distinguishing the target of interest from the dense background of endogenous proteins. Rather than relying on post-lysis labelling, which can introduce non-specific binding and high background, the most robust strategy is to label the target within the living cell prior to lysis.
This approach offers several technical advantages for subsequent lysate analysis:
- Native folding and PTMs: The protein is synthesised and processed by native cellular machinery. This ensures that PTMs and chaperone-assisted folding are preserved, factors that are often lost or altered during recombinant protein expression and purification.
- Stoichiometric precision: Covalent labelling via genetic tags ensures a 1:1 ratio of fluorophore to tag. This is critical for downstream smFRET and FCCS calculations, where knowing the labelling stoichiometry is essential for accurate distance and interaction measurements.
- Background suppression: By labelling in the intact cell and performing a subsequent wash step, the concentration of free (unbound) dye is significantly reduced before the sample is even lysed. This effectively increases the signal-to-background ratio once the sample is analysed.
Recent work from the Twelvetrees Lab (University of Sheffield) on Kinesin-1 demonstrates the efficacy of this in-cell labelling workflow. By expressing the motor protein with orthogonal genetic tags, (using self-labelling enzymes with CLIP and SNAP tags), the researchers introduced cell-permeable fluorescent ligands directly to the culture media1. In another paper by the same group, a universal protein ladder for FRET was labelled in the method described above, but also using non-canonical amino acid (ncAA) labelling2.

Figure 1 – In-cell labelling for smFRET using self-labelling enzymes and ncAAs
A) A schematic of the CLIP-SNAP fusion protein, detailing the sites that will be labelled by TMR-Star and 647 SiR, respectively
B) The workflow for in-cell labelling of HEK293 cells using self-labelling enzymes
C) Schematic of CLIP-SNAP fusion protein, detailing the sites for fluorescent labelling and the glutamine residue at position 149 for site-directed mutagenesis
D) The workflow for ncAA labelling of 293T cells
Modified figure from publications by Smith et al.1,2
How can data be collected through the noise?
In a crude lysate or serum sample, the noise is multifaceted: several components may be autofluorescent, some molecules will be incompletely labelled (donor-only or acceptor-only species in smFRET), and despite best efforts, free-floating dye will persist. Standard FRET measurements often struggle to distinguish a true, dual-labelled complex from a random burst of background fluorescence or a bright, autofluorescent vesicle.
One method to filter through this noise is to use Alternating Laser EXcitation (ALEX). It rapidly alternates between the donor-excitation and acceptor-excitation lasers (on a microsecond or millisecond timescale). This gives us two crucial parameters for every single molecule that passes through the focal volume:
- FRET Efficiency (E): Tells us about the distance between the probes
- Stoichiometry (S): Tells us if both the donor and the acceptor are present and active on that specific molecule
A significant portion of the signal might come from donor-only molecules (where the acceptor has photobleached or didn’t bind). In a traditional FRET setup, these would skew population averages.
By using ALEX, data can be plotted on a 2D E-S Histogram, permitting the selection of molecules with a stoichiometry of ~0.5 (denoting that both a donor and acceptor dye are present on the same molecule). By ignoring the clusters at S=0 or S=1, we essentially perform digital purification. This minimises the artefacts of the crude sample, leaving behind a dataset of active, dual-labelled complexes without ever having to run a single chromatography column.

Figure 2 – FRET efficiency versus stoichiometry plots with and without filtering for doubly-labelled molecules
Left – Plot contains all bursts, including donor-only molecules (S=1) and acceptor-only molecules (S=0)
Right – Plot contains only bursts from doubly-labelled molecules (S=0.5)
Crucially, this also helps filter out autofluorescence; while a signal might appear in the donor channel, it is highly unlikely to show the specific dual-excitation signature of a FRET pair.
Furthermore, ALEX provides the necessary data to calculate accurate FRET correction factors (such as γ and β). These factors account for differences in detection efficiencies and fluorophore quantum yields, which are often shifted by the complex refractive index and chemical environment of a lysate or serum sample. By applying these corrections, we transform a heterogeneous biological sample into a high-precision dataset, ensuring that the dynamics we observe are genuine.
To read more on this topic, see our technical note on capturing accurate single-molecule FRET measurements on the EI-FLEX platform.
While ALEX helps sort individual bursts in smFRET, FCS and FCCS require a different analytical method to handle the fluorescent background of a lysate. In crude media, the presence of autofluorescent species or non-specific free dye can reduce the correlation amplitude, leading to inaccurate concentration and stoichiometry measurements. To counter this, a background correction factor can be used. By measuring the fluorescence intensity of a ‘blank’ lysate (non-labelled cells) or serum sample, the contribution of the background can be subtracted from the total signal. This ensures that the calculated diffusion and cross-correlation values reflect the fluorescence of the labelled biomolecule(s) only.
Case studies
The following case studies demonstrate how researchers have successfully navigated the complexity of in-cell labelling and analysis of lysates to perform single-molecule experiments. From establishing universal benchmarks for FRET to mapping the assembly of RNA-protein complexes, these examples show that high-quality data is achievable even if the samples are considered ‘messy’.
In-cell labelling for smFRET
As discussed above, two examples that have recently been published by the Twelvetrees lab at the University of Sheffield showcase how in-cell labelling and direct analysis of cell lysates can achieve excellent smFRET data. Smith et al. developed a universal protein ladder for FRET, utilising both self-labelling enzymes and non-canonical amino acid labelling with SNAP and CLIP tags for in-cell labelling of the constructs2. FRET efficiencies between proteins that had been purified or analysed directly in cell lysates were almost identical. Another publication from the same lab demonstrated in-cell labelling of mammalian Kinesin-1, where smFRET was performed directly on diluted cell lysates in the presence of NaCl or unlabelled microtubules to observe their effect on motor protein conformational heterogeneity and autoinhibition1.

Figure 3 – FRET efficiencies are comparable between cell lysates and purified proteins from 293T cells.
Figure taken from Smith et al., 20262.
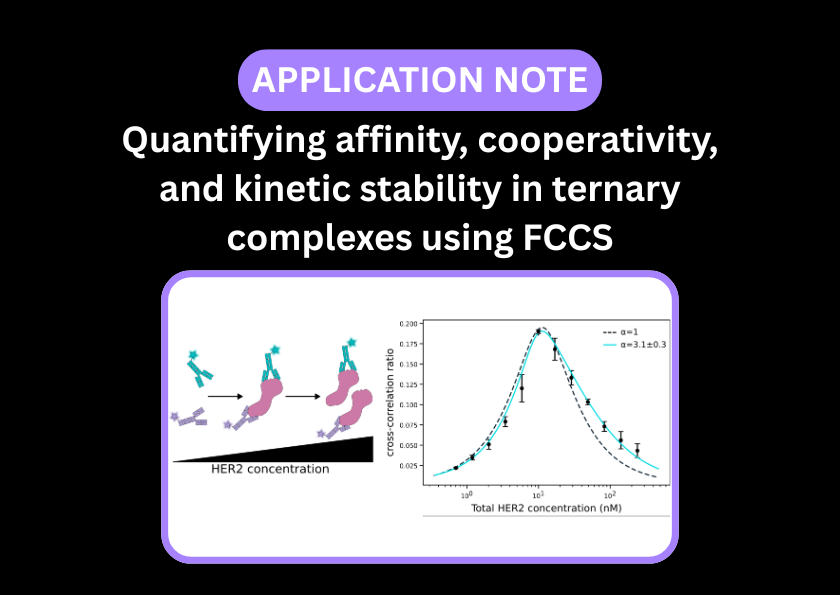
Quantifying cell-free protein production and affinity screening
In a 2025 study from the Coleman Lab (LLNL), Liu et al. developed a ‘one-pot’ system using cell-free lysates to investigate affinity screening of anti-GFP antibodies to GFP3. Instead of expressing, purifying, and then titrating GFP, they synthesised it directly in the presence of its antibody within the lysate. They used FCS to track the increasing concentration of the synthesised protein and its binding kinetics in real-time. This allowed them to generate binding curves and calculate high-precision Kd values, down to the high picomolar range, in under a few hours.
The Coleman lab performed a similar experiment using the EI-FLEX – see the application note here.
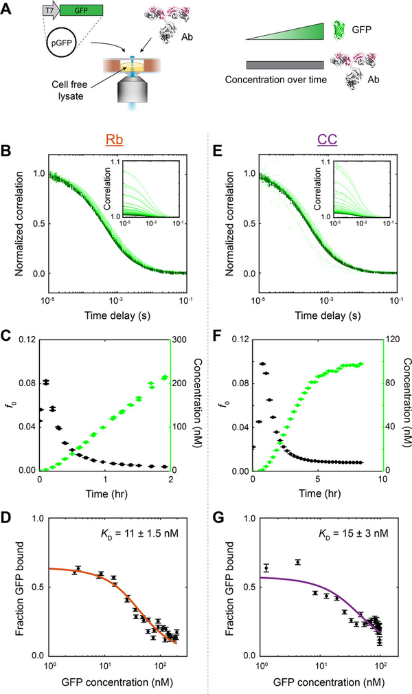
Figure 4 – FCS captures cell-free expression of GFP and antibody binding affinity
A) Experimental set-up
B) and E) Normalised correlation for Rb (commercial cell-free lysate) and CC (homemade cell-free lysate) shows that curves shift left as GFP production increases.
C) and F) Total amplitude (black) and GFP concentration (green) plotted against time for Rb and CC conditions, respectively
D) and G) Binding affinities for anti-GFP antibodies for Rb and CC conditions, respectively.
Figure taken from Liu et al., 20253.
Unveiling mRNP composition with FCS and FCCS
This study from Mateu-Regué et al. demonstrates how lysates may be favourable over live-cell imaging for complex assembly analysis of messenger Ribonucleoprotein (mRNP) complexes4. The team utilised FCS to resolve the stoichiometry of core proteins like IMP1 on single mRNA strands and FCCS to map co-diffusion to understand which factors are interacting.
While live-cell analysis captures information about trafficking and signalling events, the authors argue that cell lysates produced superior data for investigating stoichiometry, diffusion and protein interactions. Lysates could also be diluted to optimise the concentration of fluorescent particles. Crucially, the lysate format allowed for biochemical perturbations, such as controlled RNase treatments and different buffer conditions, that are technically prohibitive in a living cell. This case study demonstrates that for multi-component complexes, the messy lysate may be more conducive to quantitative discovery.

Figure 5 – Comparison of FCS data from live cell imaging and cell lysates
Normalised autocorrelation curves of for FCS data collected from free GFP (green), GFP-IMP1, an RNA-binding protein that associates with the mRNP complex, (blue) and a mutant of IMP1 that can no longer bind to RNA (red).
Both live cell FCS (top) and cell lysate FCS (bottom) showed comparable data; GFP-IMP1 demonstrated slower diffusion due to incorporation into the mRNP complex, while mutation of its RNA-binding ability results in a diffusion speed similar to free GFP.
Diffusion of proteins in cell lysates was faster overall due to the reduction of viscosity, and showed greater reproducibility and simplicity compared to live cell FCS.
Conclusion: Embracing the Chaos
With the advancement of biophysical technology, we no longer have to sacrifice biological relevance for technical precision. As these case studies demonstrate, the messy middle of cell lysates and serum is not only possible to analyse but often yields richer information than cell imaging or purified molecules. By combining strategic in-cell labelling with the digital purification of ALEX and background correction, we can observe biomolecules in their native habitat without the constraints of live-cell imaging. The ‘spherical cow’ of purified biophysics is still deeply important for a highly precise understanding of molecular structure and movement. However, to relate this precision to the living world, we need to embrace the chaotic complexity of biology.
References
- Smith, E. R., Turner, E. D., Abdelhamid, M. A. S., Craggs, T. D. & Twelvetrees, A. E. Kinesin-1 is highly flexible and adopts an open conformation in the absence of cargo. iScience https://doi.org/10.1016/j.isci.2026.114875 (2026) doi:10.1016/j.isci.2026.114875.
- Smith, E. R. et al. A Universal Protein Ladder for Standardization of Diverse FRET Assays. Advanced Science, e76672 (2026).
- Liu, C. et al. Real-Time Affinity Measurements of Proteins Synthesized in Cell-Free Lysate Using Fluorescence Correlation Spectroscopy. Anal. Chem. 97, 9638–9647 (2025).
- Mateu-Regué, À., Christiansen, J., Bagger, F. O., Hellriegel, C. & Nielsen, F. C. Unveiling mRNP composition by fluorescence correlation and cross-correlation spectroscopy using cell lysates. Nucleic Acids Res. 49, e119–e119 (2021).