In our previous article, ‘Adding dynamics to structural biology’, we established that bringing static, atomic-resolution structures to life is key to understanding the biomolecules that we are investigating. Biology happens in motion, and therefore, our experiments should capture that.
However, this adds another layer of complexity. When a modern structural biochemist or drug discovery team stands at the bench, they now have to map a static structure (from scratch or using existing data and modelling), while also choosing the right tool to capture that structure’s moving parts and binding partners.
What techniques should you combine to answer your biological questions? And when is single-molecule dynamics not the answer?
This practical guide is intended to help you answer those questions, with published case studies to demonstrate how others have utilised these complementary techniques in their own work. We analyse the experimental boundaries and opportunities of combining single-molecule Förster Resonance Energy Transfer (smFRET) with the three pillars of classical structural biology: Cryo-EM, X-ray crystallography and NMR.
smFRET and Cryo-EM
The workflow integration: Translating density to dynamics
Cryo-EM is exceptional at resolving major structural conformations or stable states. As an inherently static snapshot of a frozen population, it reveals the states that existed right before vitrification. This misses the kinetic rate constants, transition pathways, or real-time lifetimes linking those states together. Furthermore, rare or highly transient intermediate conformations often disappear during the image-classification process because they lack the particle numbers required to achieve high-resolution alignment.
To bridge this gap, structural biologists increasingly couple Cryo-EM with single-molecule FRET. By tracking distances between a donor and acceptor fluorophore, smFRET animates the static architecture provided by Cryo-EM.
Depending on the biological question, this integration can utilise distinct experimental modalities, such as solution-phase smFRET or surface-immobilised FRET.
Adding solution-phase (confocal) smFRET to Cryo-EM
Here, molecules freely diffuse through a focused laser volume. This allows researchers to capture fast, sub-millisecond equilibrium dynamics in solution completely unhindered by surface effects. This is the technique used by the benchtop EI-FLEX system at Exciting Instruments.
In their 2025 paper, Doxsey and Shen used confocal smFRET on the EI-FLEX to capture various global conformations of the Rag GTPase heterodimer1. By tracking individual heterodimers, they revealed that the subunits explored a range of static and dynamic conformations that were strictly dictated by nucleotide loading, mutations, and regulatory complexes. Confocal smFRET was ideal here, as burst variance analysis and Hidden Markov modelling found that these dynamic interconversions were occurring on a sub-millisecond to millisecond timescale.

Figure 1 – Binding of GDP alone or with GTP induces global conformational changes in Rag GTPase
Top row: smFRET histograms and FRET efficiency vs stoichiometry plots
Bottom row: Burst variance analysis, showing theoretical standard deviation of FRET efficiencies (dotted line) and experimentally observed standard deviation (triangles)
A & D) Unbound Rag GTPase (EDTA only)
B & E) Rag GTPase with GDP only
C & F) Rag GTPase with GDP and GTP
Figure adapted from Doxsey and Shen, 20251.
Adding surface-immobilised (TIRF) smFRET to Cryo-EM
Molecules are anchored to a glass coverslip (such as by biotin-streptavidin) and imaged using Total Internal Reflection Fluorescence (TIRF) microscopy. This allows individual molecules to be tracked for seconds or minutes, making it ideal for monitoring slow, processive, multi-step molecular pathways or recording extended kinetic trajectories, such as receptor signalling.
In a study examining how the pathogen Listeria monocytogenes hijacks the human MET receptor, Li et al. paired structural models and molecular dynamics simulations with in situ TIRF-smFRET directly on cell membranes2. While cryo-EM models predicted potential configurations of the active signalling dimer, TIRF-smFRET provided the experimental confirmation required to determine the precise spatial organisation of the signalling dimer.
smFRET and X-Ray Crystallography
The workflow integration: Validating lattice environments
X-ray crystallography represents the gold standard for delivering unmatched, sub-1.5 Angstrom (Å) atomic precision of well-ordered biomolecules. However, to diffract effectively, billions of proteins must pack into a highly dense, rigid, and repeating 3D crystalline lattice. This artificial environment forces the molecule to adopt a single, energy-minimised conformation, meaning highly flexible loops, allosteric domains, or transient states are more challenging to capture accurately.
smFRET can add dynamics to these static structures. In confocal smFRET, molecules freely diffuse through a focused laser volume, allowing researchers to evaluate the solution-phase behaviour of proteins in standard physiological buffers, variable salt screens, or viscous crowded environments unhindered by lattice constraints.
Adding solution-phase (confocal) smFRET to X-Ray Crystallography
Uncovering novel, dynamic conformations
Howard et al. used confocal smFRET on the EI-FLEX to explore the conformational plasticity of the bacterial accessory helicase Rep3. Historically, X-ray crystallography had identified two discrete configurations of Rep: a functional open structure and a closed structure.
The authors uncovered two previously unknown transient intermediate conformations (S2 and S3), demonstrating the influence of DNA binding and salt concentration on the interconversion rates between the four identified conformations.
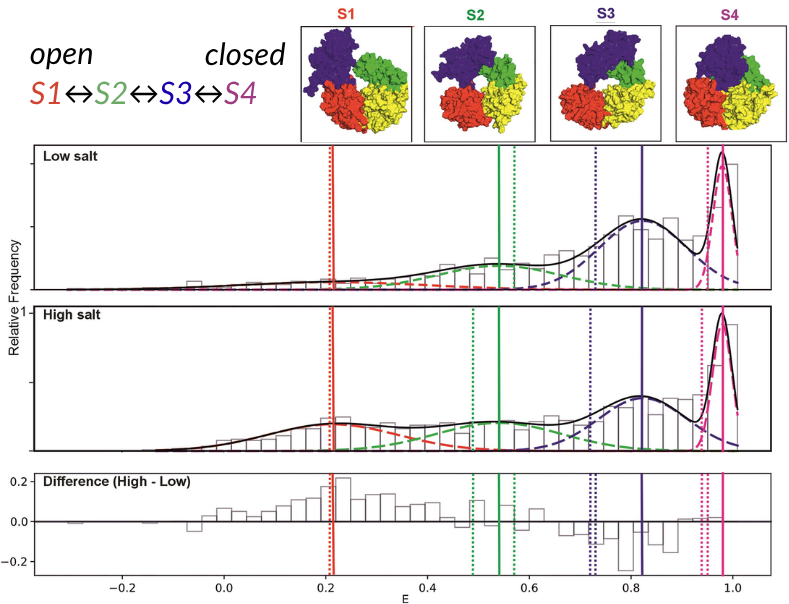
Figure 2 – FRET efficiency histograms comparing low and high salt conditions
Upper) 10 mM NaCl, Middle) 500 nM NaCl, and lower) The difference in FRET efficiencies between high and low salt conditions.
Low to high FRET efficiencies are separated into four distinct states: S1 (red), S2 (green), S3 (blue), S4 (pink).
Figure adapted from Howard et al., 20263.
Confirming X-ray crystallography-derived structures
In another paper using data from the EI-FLEX, Chadda et al. used smFRET to clarify a long-standing structural biology debate regarding Replication Protein A (RPA) – a crucial hub that shields single-stranded DNA (ssDNA) during repair4. Biochemical models suggested that RPA stretched ssDNA out linearly, while X-ray crystal structures implied ssDNA partially wrapped around the contour of RPA’s DNA-binding domains.
By measuring the exact end-to-end distances of ssDNA via confocal smFRET on the EI-FLEX, the authors found only a systematic, subtle 3 nm change upon binding. This validated that the partial wrapping configuration seen in the X-ray crystal structures perfectly reflects how the complex behaves in solution.
smFRET and NMR Spectroscopy
The workflow integration: Connecting local fluctuations to global dynamics
Nuclear Magnetic Resonance (NMR) spectroscopy has long served as a premium tool for evaluating biomolecular behaviour. It supplies high-resolution maps detailing local chemical environments, backbone flexibility, and transient hydrogen-bonding networks on microsecond-to-millisecond timescales. However, NMR faces strict physical limitations when tasked with tracking long-range global geometry or mapping large-scale domain motions, such as those observed in intrinsically disordered proteins (IDPs) or highly flexible strands of RNA.
Challenges are also encountered when analysing larger proteins or complexes, while the requirement for highly concentrated samples may inadvertently trigger massive self-aggregation or non-native oligomerisation in sticky targets.
smFRET is an ideal, orthogonal technique in these situations. It can capture conformational changes between 3 and 10 nm in length, including those in larger proteins or complexes, and the ultra-dilute picomolar working range ensures only individual molecules are analysed. The addition of fluorescence correlation spectroscopy (FCS) on the EI-FLEX can also identify aggregation, if that is a concern.
Determining the structural ensemble of a disordered protein
Naudi-Fabra et al., developed an integrated algorithm (ASTEROIDS) to model the disordered domain of the measles virus phosphoprotein5. While NMR provided excellent, fine-grained details regarding local residue-level preferences and secondary-structure propensities, it lacked the spatial reach to determine how the overall polymer chain packed together.
By incorporating a comprehensive matrix of single-molecule FRET efficiency distributions and fluorescence lifetimes into their calculations alongside NMR and SAXS data, the authors established a structural ensemble. smFRET supported the conclusion that the viral protein does not act as a randomised chain, but instead adopts a compact, highly coordinated structural layout containing functional, transiently folded motifs.
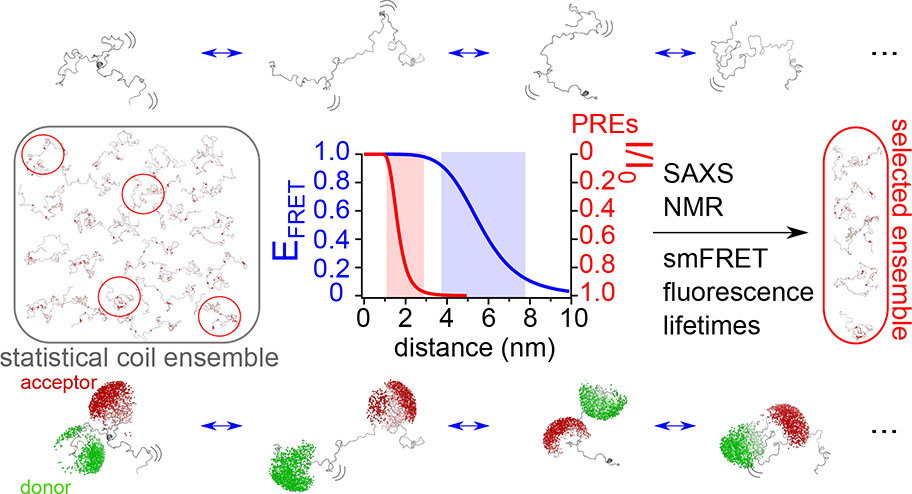
Figure 3 – Combining smFRET, SAXS, NMR and lifetime fluorescence to map IDP structures
By combining NMR and smFRET, alongside other structural and biophysical techniques, a predicted set of conformations undertaken by the IDP measles virus phosphoprotein could be derived.
Resolving RNA base-pairing patterns and long-range riboswitch folds
RNA folding poses a unique challenge; it is highly negatively charged, and therefore its global configuration is incredibly sensitive to its ionic environment, requiring techniques that can capture this structural volatility.
In a methodological framework, Hengesbach et al. demonstrated how to link NMR spectroscopy and single-molecule FRET to analyse the structural dynamics of an adenine-responsive riboswitch6. They used NMR to resolve nucleotide-resolution base-pairing patterns and local secondary structure switches, while TIRF-smFRET mapped the long-range, tertiary docking kinetics of the riboswitch’s aptamer domain over extended timelines.
What are the sample requirements and practicalities for each technique?
When deciding whether to integrate multiple techniques, it is important to consider their practicalities, in terms of experimental time, sample requirements and your biological question.
Confocal single-molecule FRET (smFRET)
- When to use: You have an existing structural model (or AlphaFold scaffold) and need to map structural dynamics, discover short-lived intermediate states, and resolve heterogeneity within the same sample
- Not ideal when: You are dealing with an entirely unknown target and require a de novo 3D atomic map from scratch, or your protein cannot tolerate any site-specific mutations/chemical dye attachments
Sample requirements:
- Concentration: Ultra-dilute picomolar regime (10-100 pM)
- Total mass: < 1 nanogram per run
- Modifications: Requires site-specific introduction of donor/acceptor fluorophore pairs (typically via self-labelling enzymes, engineered cysteines or click chemistry)
- Environment: Highly flexible. Thrives in variable salt concentrations, cell lysates or serum, viscous molecular crowding agents, and membrane-like structures, such as nanodiscs
Cryogenic electron microscopy (Cryo-EM)
- When to use: You are targeting large complexes (>50 kDa), multi-subunit assemblies, or membrane-embedded receptors in detergent/nanodiscs, and you need to sort them computationally into distinct structural classes
- Not ideal when: Your target is small (<40 kDa), highly flexible, or inherently disordered (which causes particle alignment algorithms to fail), or your sample dynamically switches states faster than the millisecond timescale
Sample requirements:
- Concentration: High micromolar regime (0.1 to 5 mg/mL)
- Total mass: 10 to 50 µg per grid trial
- Modifications: None – images the native, unmodified wild-type protein
- Environment: Extremely restricted. The sample must survive rapid vitrification (flash-freezing) in liquid ethane on a carbon-coated grid. It is highly sensitive to buffer additives like glycerol or high salt, which degrade image contrast
X-Ray crystallography
- When to use: You require an ultra-high-resolution snapshot (<1.5 Å) of a stable, rigid protein target, particularly for identifying small-molecule drug configurations inside a defined binding pocket
- Not ideal when: Your protein contains highly flexible, unstructured domains or is an IDP (flexible targets will actively prevent crystal nucleation), or you need to observe functional movements
Sample requirements:
- Concentration: Massive concentrations required (5 to 20 mg/mL).
- Total mass: Milligrams of highly purified material
- Modifications: None for native data, though heavy-atom or selenomethionine derivatisation may be required for de novo phase determination
- Environment: The protein must pack tightly into a repeating, solid 3D crystal lattice, surrounded by volatile crystallisation liquors containing heavy precipitants, salts, or organic solvents
NMR spectroscopy
- When to use: You are profiling small-to-midsize targets (<50 kDa), highly flexible intrinsically disordered proteins (IDPs), or nucleic acids, and you require residue-specific information on local side-chain habits, hydrogen bonding, or local fast dynamics (ns-µs)
- Not ideal when: Your target is large (>100 kDa), or your protein precipitates out of solution at high concentrations
Sample requirements:
- Concentration: High micromolar to millimolar levels (50 µM to 1 mM)
- Total mass: 1 to 10 mg of pure material per NMR tube
- Modifications: Demands expensive isotopic labelling, requiring protein expression in restrictive media
- Environment: Requires high sample stability over days of continuous scanning, specific non-interfering buffers, and typically a fraction of deuterium oxide for signal locking
Summary – Orchestrating multi-technique workflows
Pairing structural methods with an orthogonal technique unlocks answers that neither could achieve in isolation. The table below outlines the biophysical combinations we have discussed, when to deploy them, and how they work together.
To learn more about what other techniques are complemented by confocal smFRET beyond those discussed in this article, see our technology page.
Combination | Biological objective | Workflow | Benefits |
Cryo-EM + Confocal smFRET | Mapping high-resolution transient states | 1. Resolve major 3D structural classes via Cryo-EM 2. Use confocal smFRET to track free-diffusion solution kinetics across those same domains at room temperature | Expands static 3D density maps into a dynamic structural landscape – capturing intermediate states that grid vitrification or computational smoothing might have missed |
NMR Spectroscopy + Confocal smFRET | Modelling intrinsically disordered proteins (IDPs) or highly flexible complexes | 1. Use NMR to gather fine-grained, residue-specific local backbone habits and secondary structure propensities 2. Use smFRET to provide long-range (3-10 nm) global distance constraints | Prevents computational structural models from drifting, proving whether a disordered chain adopts a completely randomised structure or a compact, functional native layout |
X-Ray Crystallography + Confocal smFRET | Eliminating crystal lattice constraints for structural validation | 1. Use an existing or novel PDB crystal model to locate rigid domain configurations or flexible loops 2. Place fluorophores flanking those regions to screen the target at picomolar levels in physiological liquid buffers | Identifies and removes crystal-packing bias, proving whether a crystallised conformation represents a true biological state or an artefact of forced lattice packing |
Cryo-EM + TIRF-smFRET | Tracking multi-subunit assembly and processive action on surfaces | 1. Map out the 3D architecture of massive macromolecular machines or membrane-bound complexes using Cryo-EM. 2. Anchor the complex to a surface to track slow, processive, multi-step pathways or lateral assembly over seconds/minutes | Connects large-scale cellular structures directly to functional, long-lived mechanical timelines (such as processive viral replication or ribosome translocation steps) |
References
- Doxsey, D. D. & Shen, K. Global conformation of the Rag GTPase heterodimer governs eukaryotic amino acid sensing. Proceedings of the National Academy of Sciences 122, e2517050122 (2025).
- Li, Y. et al. Single-molecule imaging and molecular dynamics simulations reveal early activation of the MET receptor in situ. bioRxiv 2023.12.22.572978 (2024) doi:10.1101/2023.12.22.572978.
- Howard, J. A. L. et al. The transitional kinetics between open and closed Rep structures can be tuned by salt via two intermediate states. Nucleic Acids Res. 54, gkaf1483 (2026).
- Chadda, R. et al. Partial wrapping of single-stranded DNA by replication protein A and modulation through phosphorylation. Nucleic Acids Res. 52, 11626–11640 (2024).
- Naudi-Fabra, S., Tengo, M., Jensen, M. R., Blackledge, M. & Milles, S. Quantitative Description of Intrinsically Disordered Proteins Using Single-Molecule FRET, NMR, and SAXS. J. Am. Chem. Soc. 143, 20109–20121 (2021).
- Bains, J. K. et al. Combined smFRET and NMR analysis of riboswitch structural dynamics. Methods 153, 22–34 (2019).


